
#NephJC Chat
Tuesday May 8th 9 pm Eastern
Wednesday May 9th 8 pm BST, 12 noon Pacific
J Am Soc Nephrol. 2018 Apr 13. pii: ASN.2017111200. doi: 10.1681/ASN.2017111200. [Epub ahead of print]
NPHP1 (Nephrocystin-1) Gene Deletions Cause Adult-Onset ESRD.
Snoek R, van Setten J, Keating BJ, Israni AK, Jacobson PA, Oetting WS, Matas AJ, Mannon RB, Zhang Z, Zhang W, Hao K, Murphy B, Reindl-Schwaighofer R, Heinzl A, Oberbauer R, Viklicky O, Conlon PJ, Stapleton CP, Bakker SJL, Snieder H, Peters EDJ, van der Zwaag B, Knoers NVAM, de Borst MH, van Eerde AM.
PMID: 29654215 Free Full Text thanks to JASN



Introduction
To most Nephrologists, Nephronophthisis (NPH) is considered to be a rare autosomal recessive paediatric disease. However, this article by Snoek et al challenges that belief, by examining adults with end-stage kidney disease (ESKD) and looking at the prevalence of mutations that cause NPH.
Background
NPH is the most prevalent genetic cause of ESKD in children, accounting for 15% of paediatric ESKD. Approximately 25% of children with NPH harbor homozygous deletions in the NPHP1 gene that encodes the Nephrocystin-1 protein, leading to ESKD at a median 13 years of age (OMIM number 607100). Nineteen other genes are associated with NPH. The disease is autosomal recessive inheritance pattern, so heterozygotes do not develop NPH or ESKD (For a review see Srivastava & Sayer).
The diagnosis is typically made several years after the onset of symptoms. Most patients do not have extra-renal manifestations of NPH. The clinical presentation usually includes polydipsia, polyuria, growth retardation and anaemia. Corticomedullary cysts are seen in half of NPH patients. Hypertension is typically absent due to the salt wasting nature of the disease. Genetic testing is the only reliable method to diagnose NPH. Kidney biopsy only reveals non-specific features of advanced CKD.
About 10% of NPH patients get classified into clinical syndromes based on the presence of extrarenal manifestations. These include:
Joubert syndrome
Bardet-Biedl syndrome
COACH syndrome
Jeune syndrome
Meckel-Gruber syndrome
Senior-Løken syndrome
Leber congenital amaurosis
oculomotor apraxia, Cogan type.
NPH is considered to be a ciliopathy, because the genes involved in NPH encode proteins of the primary cilium, a sensory organelle. This explains the extra-renal manifestations present in the syndromic forms of NPH including retinal degeneration, cerebellar ataxia, liver fibrosis and situs inversus.
No specific treatment exists for NPH. Management includes correction of electrolyte anomalies, adequate hydration and treatment of the complications of CKD. Some extra-renal manifestations can be treated.
With only sporadic case reports in the literature, very little is known about NPH in adults. This study goes some way to filling that knowledge gap by looking at the prevalence of NPHP1 deletions in adults with ESRD.
Methods
5606 adults with ESKD, who later received a renal transplant, were identified from the iGeneTRAiN consortium, a worldwide collaboration of transplant genome-wide association studies (GWAS).
Inclusion criteria defined adult-onset ESKD. They genotyped five iGeneTRAiN Consortium cohorts, consisting of renal transplant recipients and (donor) controls.
They examined the NPHP1 gene for copy number variants (e.g. insertions/deletions) and those patients showing complete homozygous NPHP1 deletions were validated using independent polymerase chain reaction.
Interestingly, consent had not been obtained to return genomic data findings back to the participants, so the information on homozygous or heterozygous NPHP1 gene status could not be provided back to participants at an individual level.
Results
The authors found a 0.5% prevalence (n=26) of complete NPHP1 homozygous deletions within the cohort. No deletions in the other 19 known NPH genes were observed (Figure 1).

Figure 1 from Skoen et al, JASN, 2018
Conversely, none of the 3311 kidney donors studied had NPHP1 deletions, demonstrating that the deletions were restricted to those only with ESKD.
The frequency of heterozygous NPHP1 deletions was higher in those with ESKD than the kidney donor cohort (n=36 versus n=10).
The median age of ESKD in patients exhibiting NPHP1 homozygous deletions was 30 years.
Only three of the 26 NPHP1 deletion patients had been given a previous diagnosis of NPH, many others carrying a label of ESKD of unknown cause (n=11), or another diagnosis (Figure 3). Two patients exhibited extra-renal manifestations that associate with NPH including congenital blindness (Senior-Løken syndrome) and neurodevelopmental delay (Joubert syndrome). Neither patient had a previous diagnosis of NPH.
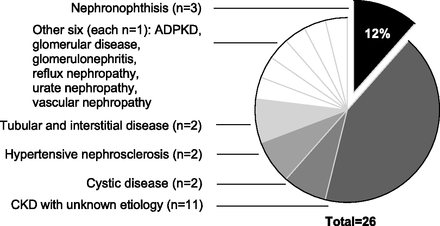
Figure 3 from Snoek et al, JASN 2018. What were the diagnosis of the patients prior to genomic analysis.
Discussion & Conclusions

Figure 4 from Skoen et al. Authors conclusions: At least one in 200 (0.5%) patients has adult-onset ESKD due to nephronophthisis. The late presentation of nephronophthisis might be due to genetic modifier effects. Accurately diagnosing a monogenic disease such as nephronophthisis can have wide-ranging clinical implications.
NPH is both a paediatric and an adult disease
Over half of this adult cohort developed ESRD at 30 years or older, far beyond the known median of 13 years in juvenile NPH. We can also see that the prevalence of NPHP1 deletions in adults with ESRD is far higher than initially thought; prior to this study only six patients were reported to have NPHP1 mutations accounting for ESKD beyond the age of 30 years. These observations support the idea that NPH should now be considered an adult as well as a paediatric disease. The reason that some NPH patients develop ESKD later in life than others is an area ripe for research.
Is the prevalence of NPH in adult ESKD really 0.5%?
This study may actually underestimate the number of adults with NPH. This study examined NPHP1 gene deletions only, and not the other 19 genes involved in NPH. It is therefore likely that the true prevalence of NPH is higher than that estimated from NPHP1 homozygous deletions alone. In keeping with this, the frequency of heterozygous NPHP1 deletions was higher in those with ESKD than the kidney donor cohort, making compound heterozygosity (a full gene deletion in one allele with a different pathogenic mutation in the other allele) a distinct possibility and one that would increase NPH number beyond those observed with NPHP1 homozygous deletions alone. Finally, smaller homozygous NPHP1 gene deletions were not examined in this study.
Can we be certain that the 0.5% of patients with NPHP1 deletions had NPH as their cause of ESKD? The answer depends upon the penetrance of NPHP1 deletions in adults. We know from children that homozygous NPHP1 deletions demonstrate complete penetrance, meaning that all those with homozygous NPHP1 deletions have NPH. It is likely that the same holds true in adults.
Can the data be extrapolated to populations outside the study cohort? To answer the question fully, the analysis would need to be performed in an independent cohort of ESKD patients. This cohort was primarily Caucasian patients. The 26 participants exhibiting NPHP1 deletions were not thought to be related, up to and including the third degree relative. This suggests that the results may be extrapolated to other populations and that the number of NPHP1 deletions observed was not influenced by an over-enriched cohort.
There is no treatment, why should I care?
Why is it important to diagnose NPH if no treatment exists? A diagnosis of NPH avoids subjecting the patient to unnecessary treatments for an erroneous diagnosis, such as potentially harmful immunosuppression used in the treatment of glomerulonephritis. It can help with decisions regarding living donors – siblings that have inherited NPH-causing mutations would be unsuitable donors.
This study illustrates a novel application of GWAS data outside of its original intended use. These GWAS studies were performed to examine the influence of genetic variants on transplant outcomes but have now been used to interrogate ESRD cause. We expect further studies to follow regarding other ESRD aetiologies. Moreover, as kidney donors are included in iGeneTRAiN, non-renal phenotypes may also be examined.
So, our questions to the adult Nephrologists are ‘How many cases of adult NPH have you unwittingly seen, and when will you consider NPH next time an adult presents with CKD or ESRD of unknown etiology?’
A warm welcome awaits you to join us in discussing these questions and more at our NephJC Twitter Journal Club. New participants are always very welcome.